Medical Device Manufacturers
How does the MDR define the broader responsibilities for manufacturers & what does this mean for the industry?
What are manufacturer’s responsibilities under the MDR?
The medical device sector is one of the most highly regulated market sectors in the world. The introduction of the EU MDR — Medical Device Regulations (MDR) 2017/745 — sees the largest overhaul of medical device legislation in decades.
All medical devices and medical device manufacturers in the European Union (and, under transitional arrangements, some devices in the UK) fall under the EU MDR 2017 / 745. The MDR governs essentially all activities from device conception and design, through realisation, manufacture, stakeholder engagement, distribution and surveillance activities once commercialised. This means that the MDR impacts all economic operators within the European medical device market.
Because the scope of the MDR is broader than that of the previous MDD legislation, manufactures familiar with the MDD may face challenges during the transition to the more extensive MDR legislative framework. Manufacturers producing new-to-market devices will need to develop an MDR compliance strategy for their product in order to obtain first approval. Either way, the MDR represents a significant challenge for many manufacturers.
In order to start working with the MDR effectively, it is important to understand the full range of responsibilities of medical device manufacturers under the MDR. Article 10 MDR outlines the general requirements for all medical device manufacturers and other economic operators under the MDR.
Article 10 requires that all manufacturers must:
- Ensure that their medical devices are designed and made in accordance with MDR
- Have a Risk Management system in accordance with Annex I Section 3 of the MDR
- Conduct an appropriate Clinical Evaluation of all their devices
- Maintain a record of up to date technical documentation
- Have access to a Person Responsible for Regulatory Compliance (PRRC)
- Have an implemented Quality Management System (QMS)
If MDR compliance has been successfully demonstrated following a suitable conformity assessment (involving a Notified Body in many cases), manufacturers are required to affix a CE-mark to their devices and must comply with requirements for using a Unique Device Identification (UDI) system.
They must also ensure that procedures are in place to ensure that device production continues to be in conformity with MDR as it scales, with any changes declared to regulators.
Let’s examine some of these manufacturer responsibilities under the MDR in detail.
Quality Management System
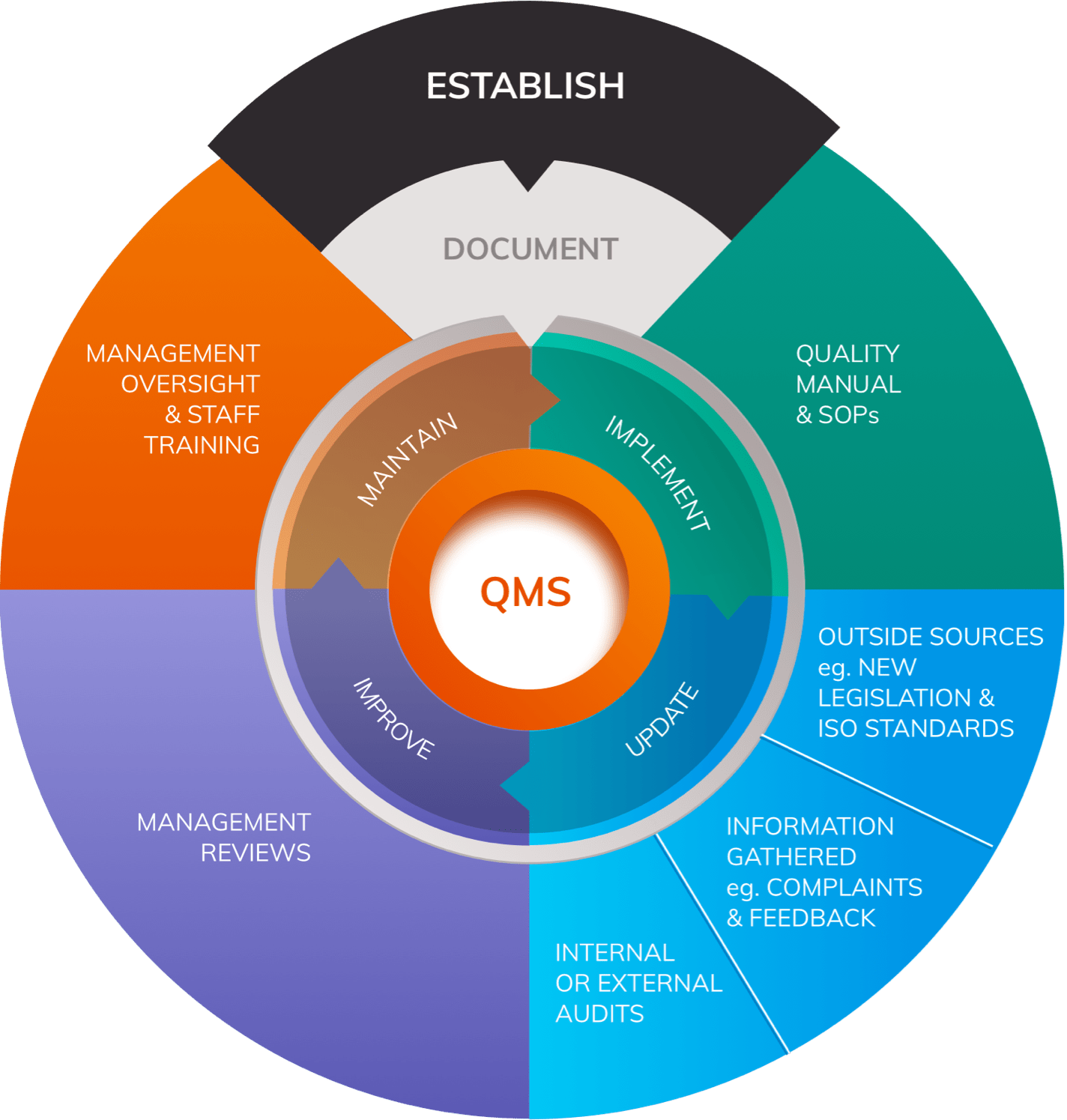
Article 10 outlines requirements for the Quality Management System (QMS) that all manufacturers must establish.
The QMS must cover all aspects of the manufacturer’s organisation, extending from basic corporate processes such as sourcing, to detailed procedures for product realisation and Post-Market Surveillance strategies.
The harmonised standard ISO 13485:2016, developed in reference to the MDD, outlines detailed requirements for quality management systems for medical devices.
It is important to understand that the MDR contains additional obligations that extend beyond those outlined in ISO 13485:2016 in certain areas. Nonetheless, the ISO standard is an important source of guidance for manufacturers when building a QMS.
Person Responsible for Regulatory Compliance (PRRC)
Article 15 MDR states that every manufacturer must have access to a dedicated Person Responsible for Regulatory Compliance. This nominated person must take overall responsibility for regulatory activities at the company, and must be allowed to undertake their duties without prejudice, even if their proper activities may result in an action that is commercially disadvantageous to the company (for example, requiring withdrawal of a device from the market on safety grounds).
Most companies will be required to employ their own responsible person. However, the MDR does make allowances for small manufacturers who need not directly employ their own person but must have one “permanently and continually” at their disposal.
Unique Device Identification System
Articles 27-29 MDR require manufacturers to ensure that their devices can be uniquely identified according to a specific, EU-wide UDI system. All devices must have a Basic UDI-DI at a regulatory level, registered in the EUDAMED database. At a product level, devices must display a UDI carrier. The MDR also requires that manufacturers of higher risk devices maintain a record of where all devices have been sold to.
Clinical Evaluation and Clinical Investigations
It is a requirement that manufacturers conduct a Clinical Evaluation of all their medical devices; a process fed in part by data generated through clinical investigations undertaken by the company. A Clinical Evaluation must be summarised in a technical document known as a Clinical Evaluation Report (CER).
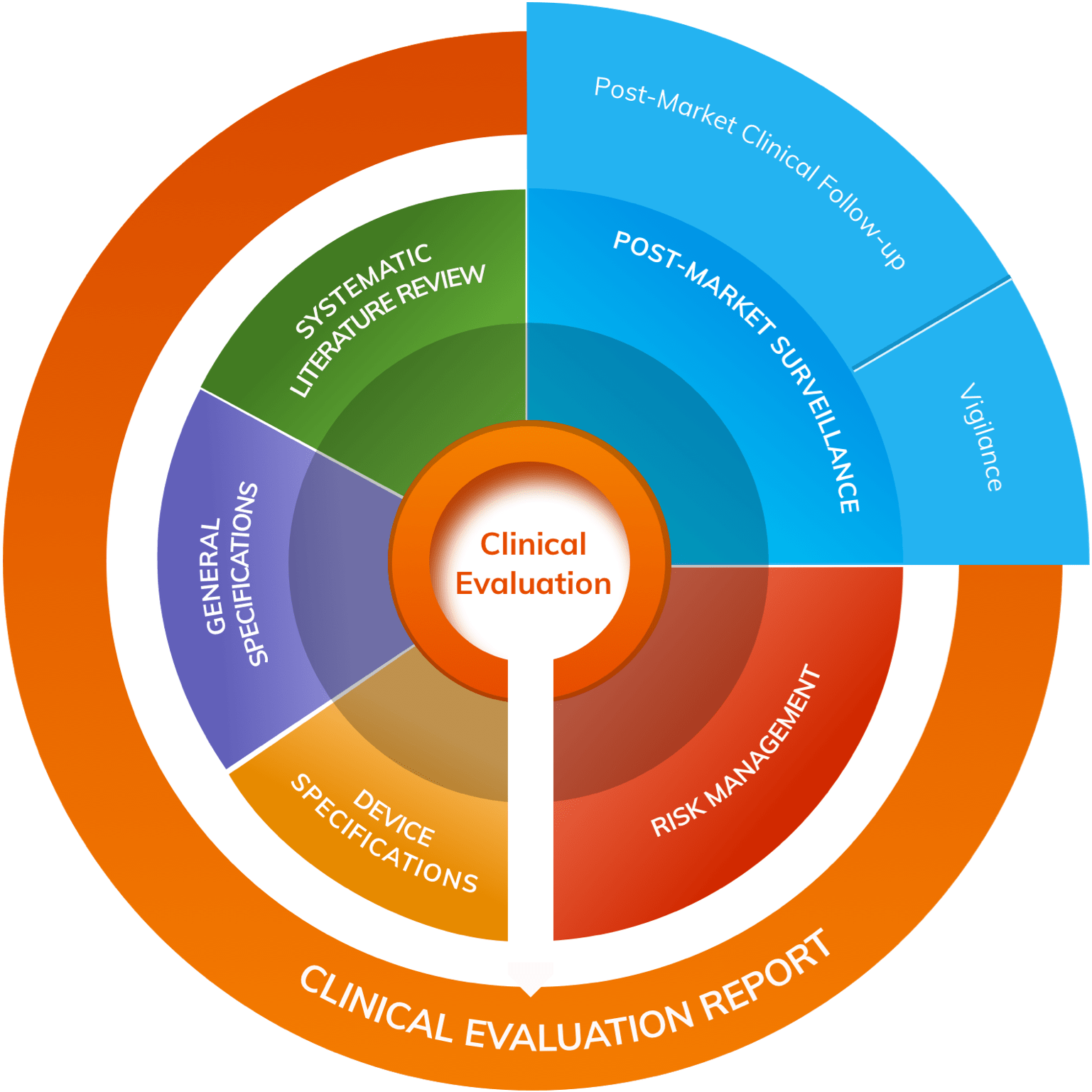
Clinical evaluations and clinical investigations are defined in Article 2 MDR as follows:
- Clinical Evaluation: Systematic and planned process to generate continuously, collect, analyse and assess the clinical data of a medical device in order to verify safety and performance.
- Clinical Investigation: Any systematic investigation involving one or more human subjects, undertaken to assess the safety or performance of a medical device.
Under MDR the stated objectives of Clinical Evaluation are to demonstrate:
- Conformity with the relevant General Safety & Performance Requirements (GSPRs)
- Suitability for intended purpose
- Alignment with safety & performance objectives
- An acceptable benefit-risk ratio
All manufacturers must plan, conduct and document a Clinical Evaluation following the process detailed in Annex XIV Part A.
Post-Market Surveillance (PMS)
Once a medical device has been released onto the market it is the responsibility of the manufacturer to constantly monitor its safety and performance through establishing and running a Post-Market Surveillance (PMS) system.
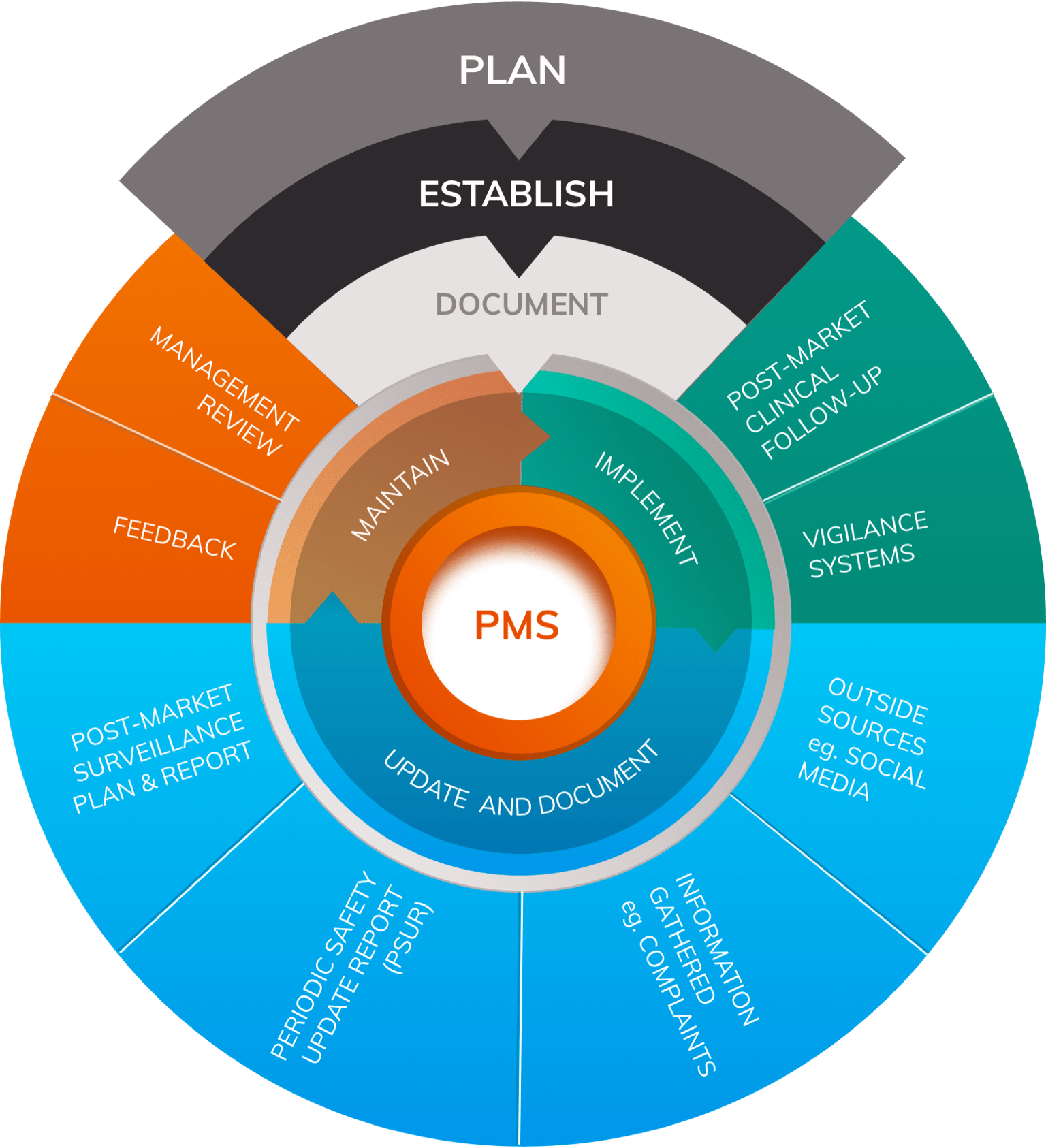
PMS, required by Article 83 MDR, is performed on a plan-implement-review-act basis and forms a key part of a manufacturer’s Quality Management System (QMS). Effective PMS systems contain multiple elements to ensure that comprehensive data is accumulated on the safety and performance of the device throughout its lifetime.
Vigilance is one arm of an effective PMS system and is ‘always open’ in order to react to events such as serious incidents or complaints. Post-Market Clinical Follow-up (PMCF) is another aspect of PMS that comprises the prospective collection of clinical data relating to the performance of the device following its release onto the market.
Market Surveillance - not to be confused with Post-Market Surveillance is a form of device performance monitoring mandated by MDR Article 93. Unlike PMS, Market Surveillance is performed by Competent Authorities rather than manufacturers. It provides a ‘top down’ perspective on the safety and performance of each class of device and also ensures that manufacturers of all devices, including self-certified Class I devices, meet all relevant MDR requirements.


