The Vigilance process in Post-Market Surveillance
The importance of vigilance as a component of medical device EU MDR PMS
What is medical device Vigilance and how does it relate to Post-Market Surveillance?
Medical device Vigilance is the process of establishing and running a system to collect and report data on serious incidents, Field Safety Corrective Actions (FSCAs), and the monitoring of trends of expected side-effects. It is a reactive process that is a crucial element of all Post-Market Surveillance (PMS) systems under the MDR.
Rules for Vigilance are set out in Articles 87 - 90 of the MDR.
How does Vigilance differ from Post-Market Surveillance and PMCF?
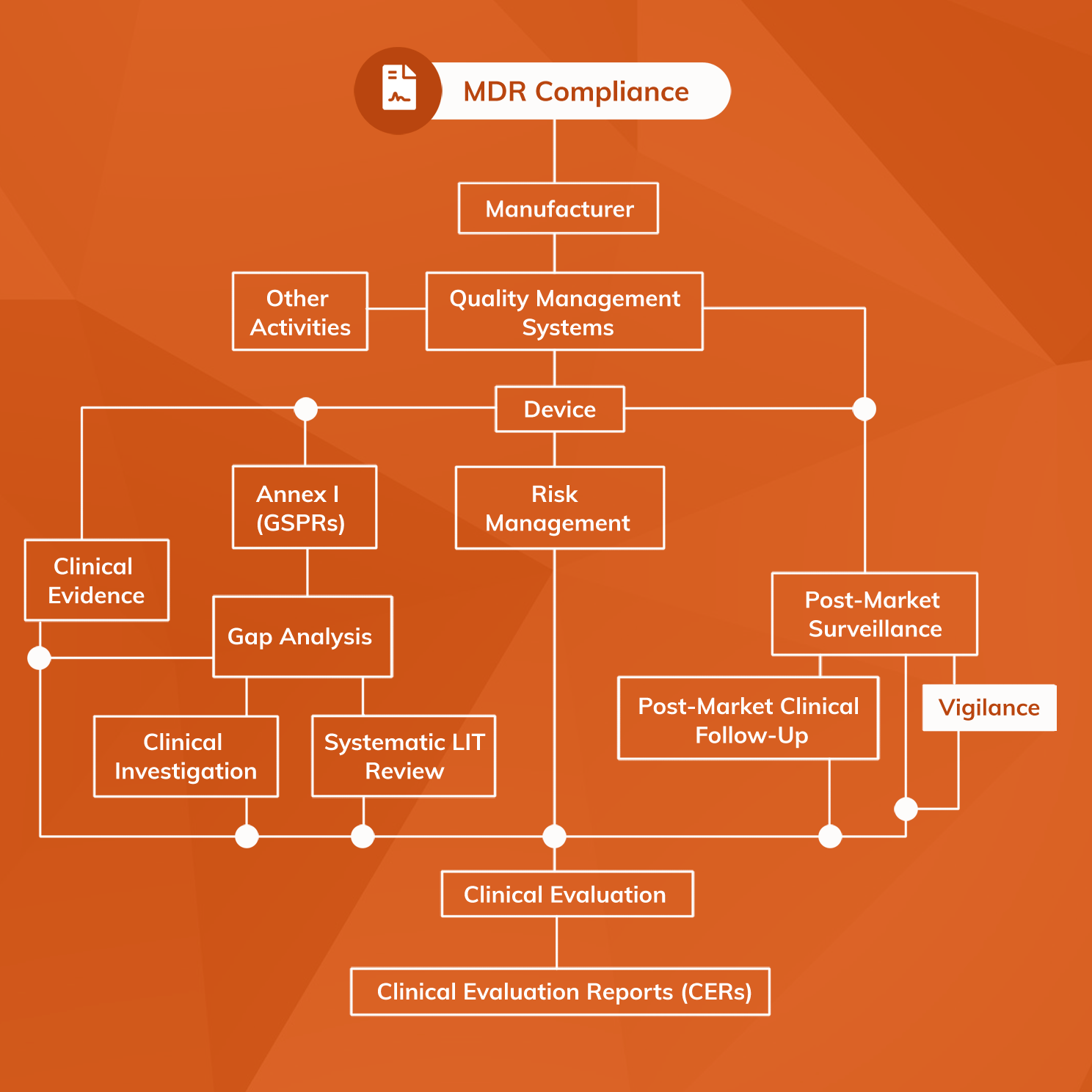
PMS, Vigilance and Post-Market Clinical Follow-up (PMCF) are easy to confuse because they all concern the collection of data on the safety and performance of CE-marked medical devices. However, while complementary and inter-dependent, they are not all the same and it is important to understand both how they differ and how they relate to one another.
PMS is a broad system that encompasses both Vigilance and PMCF. It is the overall process for monitoring and reacting to medical device performance following release onto the market.
PMCF concerns the proactive collection and analysis of clinical data through the generation of Real World Evidence (RWE) on the safety and performance of the device in normal use.
Vigilance, by contrast, requires a reactive system that is capable of collecting information in the event of a serious incident, complaint, or adverse event.
Therefore PMCF and Vigilance are components of a PMS system, which describes an overall strategy for monitoring medical device performance after release onto the market.
How is an MDR Vigilance plan developed?
A MDR Vigilance plan must be set out as a component of the PMS plan required by Article 83.
The Vigilance plan must include methods for:
- collecting data on serious incidents
- distinguishing serious incidents from expected side effects
- analysing serious incidents and procedures for escalating to competent authorities
- receiving and analysing complaints
- compiling data on trends
Article 89 requires that the Vigilance plan must describe a process for following up on serious incidents. The follow-up process must include:
- An investigation on the facts
- A risk assessment
- Communication with notified body and competent authority
- Provision of devices for examination “as is” without making any changes to them.
The Vigilance plan must also include a method for reporting significant data obtained through the Vigilance system to regulatory authorities.
What are the requirements for MDR Vigilance?
MDR Vigilance must satisfy the requirements in Articles 87 - 90 of the MDR. The system must be outlined in technical documents that form part of the portfolio required by Annex II MDR.
Manufacturers are required to report serious incidents to competent authorities as soon as a causal relationship to the device has been established, and absolutely within the following time frames:
- no later than 15 days after most serious incidents
- no later than 2 days post incident if it represents a serious threat to public health
- no later than 10 days post incident in the event of a death
- it is permissible for manufacturers to submit a partial report initially in order to meet these deadlines.
The document MedDev 2.12-1 - Guidelines on a medical device Vigilance system published by the European Commission provides detailed guidance on constructing an MDR-compliant Vigilance system.
How MDR Vigilance is different to MDD Vigilance?
MDR Vigilance is far more extensive than Vigilance under the MDD and is better differentiated from PMS. In many ways, the additional detail within the text of the MDR relating to Vigilance, compared with the incomplete description in the MDD, is a real benefit to manufacturers — under the MDR, expectations and requirements are far more clearly defined and analysing whether expectations have been met is easier and more transparent.


