Real World Evidence for medical devices
The importance of Real World Evidence (RWE) to Post-Market Clinical Follow-up and EU MDR compliance
What is Real World Evidence for medical devices?
Real World Evidence (RWE) is clinical evidence generated from observational data on the real-world use of medical devices. It differs from evidence produced through pure clinical trials because RWE relates to experience with a device in all settings and in all types of patient/user normally exposed to the device. RWE studies vary in type and scope but may run indefinitely rather than being time limited, continually adding new subjects to the dataset in the form of a registry.
RWE has become increasingly important following the enactment of the Medical Device Regulation MDR 2017/745, fully implemented from May 2021. The MDR introduces enhanced requirements for Post-Market Clinical Follow-up (PMCF) of all medical devices, requiring that data be produced continually and throughout the entire lifetime of the device.
In order to meet these requirements, medical device manufacturers need to develop and implement a Clinical Evidence strategy that produces RWE on device safety and performance, while remaining in compliance with all relevant rules, regulations and wider legislation.
What are the differences between Real World Evidence and standard clinical data?
RWE has some important differences to other forms of clinical data and requires an adapted approach that may be less familiar. “Standard” clinical data is generated through formal clinical trials that have the following general characteristics:
- Carefully controlled inclusion and exclusion criteria, meaning the study population can be limited to a sample
- Set ‘case’ and ‘control’ parameters enabling a direct comparison between two defined populations
- Often involves some form of experimental exposure to address an hypothesis
- A limit on recruitment number
- A limit on study time period
- Relatively few, often highly specialised, clinical sites at which the study is conducted.
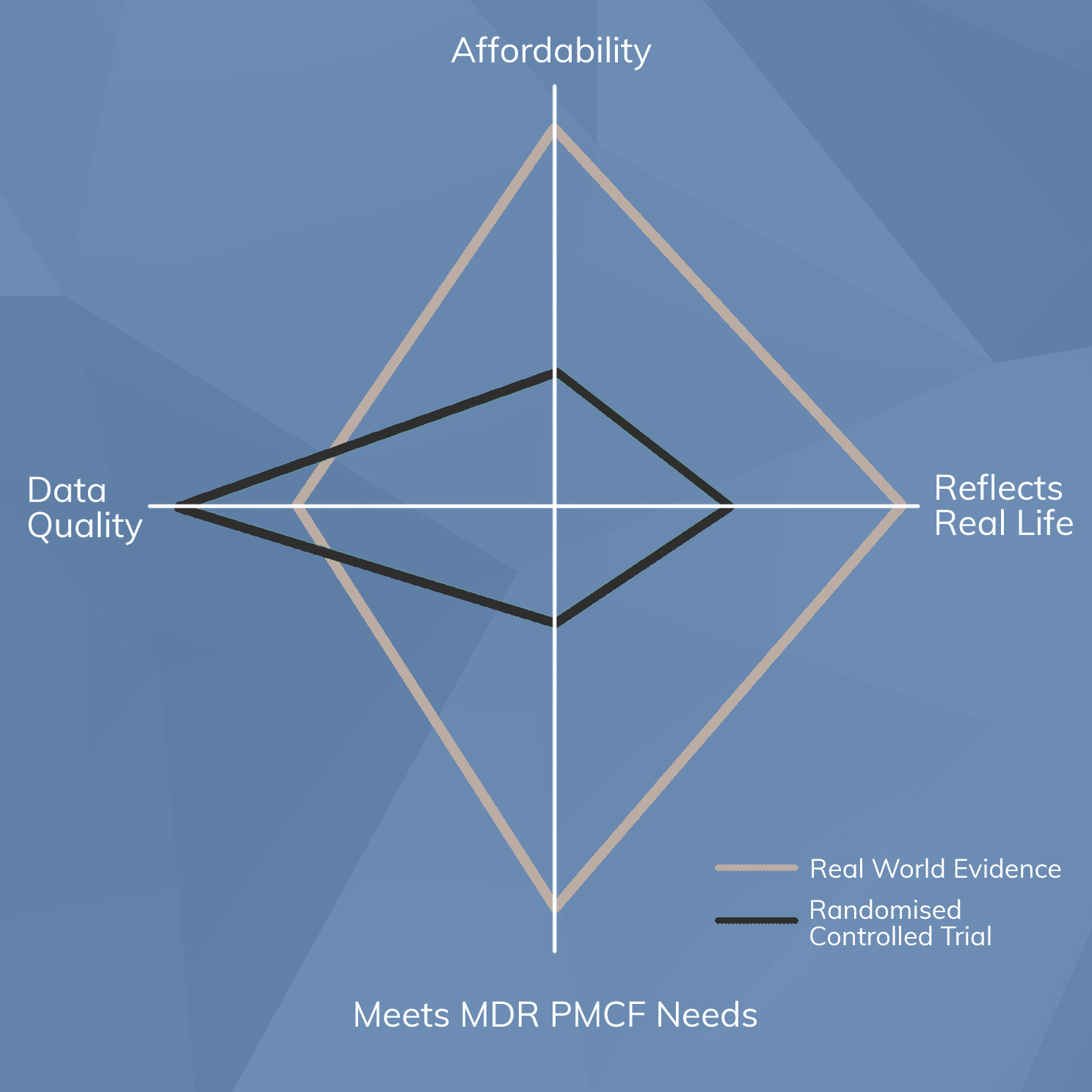
By contrast, RWE of the type required to meet the requirements for PMCF systems under the EU MDR does not conform to these characteristics. Instead, Real World Evidence:
- Is drawn from a study population that represents the entire population normally exposed to the device
- Is non-comparative, focusing only on the performance of the subject device rather than making comparisons to something else
- Does not involve experimental exposure. Instead, it studies the use of a device that is already approved for use in the manner being studied, and seeks to confirm or refute that it meets safety and performance objectives “out there in the world”.
- Has no upper limit on recruitment number
- Runs indefinitely, aiming to cover the “entire lifetime of the device”
- Is conducted at all types of clinical site, not just specialist units
From this comparison it should be clear that planning, implementing, performing, monitoring and reporting RWE is entirely different to that of “standard” investigative clinical data.
Designing a system to generate RWE while remaining compliant with all relevant rules and legislation is a demanding task that requires specialist knowledge. It begins with choosing the most appropriate method for generating RWE from the various options available.
What are the methods for Real World Evidence generation?
There are numerous different sources of RWE. The applicability and suitability of a particular source or method will vary depending upon several factors, including:
- the type of medical device
- its complexity
- its risk classification
- the depth and detail of data needed
- the number of exposed subjects or users
- existing sources of data
It is important that the most appropriate method is selected for any given device. An inappropriate choice of RWE generation method may lead to a progressive accumulation of problems, as any weaknesses in the collection method or the data itself will compound over time.
PMCF surveys
Surveys are comparatively easy to implement and are an appropriate choice for many types of device that are used by laypersons outside a formal healthcare setting. Patients or users respond to a series of standardised questions following exposure to a medical device and the data is then accumulated and analysed. Patient surveys are reliant upon subjects being motivated to respond and can be prone to bias - often, those who respond represent a more motivated cohort than average, potentially applying a positive skew to the data. Despite these shortcomings, surveys can be an appropriate design to obtain RWE on simple or low risk medical devices, or those sold directly to the public.
Retrospective cohort reviews
Retrospective cohort reviews are another reasonable but unsophisticated source of RWE. In a retrospective review, the medical records of subjects meeting specified exposure criteria are analysed in order to glean historical safety and performance data. This method has the advantage of being less prone to patchy subject compliance than patient surveys, but remains susceptible to positive selection bias because it is possible for reviewers to (unwittingly) select cohorts more likely to have positive outcomes. Eliminating bias entirely from retrospective reviews is challenging, requiring strict controls on data collection methods. Regulatory bodies will need to see evidence of stringent bias control methods in order for the data generated to be accepted as valid.
National registries
National registries represent a powerful source of RWE that is inherently less prone to bias than other methods. Where they exist, a national registry (such as the UK National Joint Registry (NJR)) collates data on every use of or exposure to a medical device in a certain category. In the case of the NJR, safety and performance data on every implantation of prosthetic hips, knees, shoulders and ankles is collected in the database, producing a massive and comprehensive source of bias-free data. Unfortunately, national registries only exist for a limited number of categories of device, and in the absence of such a national registry it may be necessary for manufacturers to build their own.
Medical Device Product Registries
Medical Device Product Registries, in the absence of a national registry, represent the gold standard source of RWE for medical devices. The design of the registry can be carefully controlled to ensure that sources of bias are eliminated, and the use of a study protocol will help to ensure that the investigation proceeds according to a prospective, specified standard at all clinical sites, eliminating the potential for variability. A properly-designed registry will be capable of capturing RWE on device performance, adverse events, emerging risks, trends of side-effects, and systematic misuse, meeting the full range of requirements for PMCF as specified in MDR Annex XIV Part B and ensuring regulatory acceptability of PMCF methods both now and into the future.
What documentation is required for Real World Evidence in the MDR?
Real World Evidence generation systems, as a component of PMCF, will need to be documented in accordance with requirements for technical documentation as specified in the MDR. Annex II and III specify a range of documents that must be produced in support of every medical device, including a PMCF Plan, PMCF Report and a Clinical Evaluation Report (CER).
Additionally, MDR Annex XV lists a range of requirements that must be adhered to for every form of clinical investigation, including those intended to generate RWE. These requirements include the need to produce a clinical investigation plan, investigator’s brochure, and informed consent form, as well as securing ethical approval where required and adhering to data protection laws and regulations.
Overall, constructing a robust system for generating Real World Evidence is a specialist task but one that will safeguard the regulatory approval status throughout its commercial lifetime.
To learn more about support available in selection and design of RWE generation systems, contact us for a free initial consultation.


