Medical Device Risk Management Process
A detailed guide on MDR-compliant Risk Management for medical devices
What is Risk Management of medical devices?
Risk Management is the structured process of identifying, analysing, controlling, and monitoring risks associated with use of a medical device. It also concerns evaluation of clinical benefits and the benefit-risk ratio on an ongoing basis. It is a fundamental component of all medical device regulatory submissions.
All medical devices have inherent risks. It is a fact of healthcare that every patient interaction carries with it a risk of harm, and the use of medical devices is in no way an exception.
Risk Management is not the process of eliminating every conceivable risk that could emanate from use of a device. Rather, it is the elimination of unacceptable risks, along with the mitigation of any risks that cannot reasonably be eliminated.
Performed correctly, Risk Management allows the formulation of a risk benefit assessment (expressed as “benefit-risk” in the text of the EU MDR) that will determine whether the potential benefits of using a device outweigh any residual risks.
What is a ‘medical device risk’?
The formal definition of a ‘medical device risk’ is provided in Article 2 MDR, where it is stated that it is:
…the combination of the probability of occurrence of harm and the severity of that harm.
Therefore, acceptability of a medical device risk and processes required to ensure its elimination or mitigation will differ according to a function of both severity and frequency.
Medical device risks must not be confused with other categories of risk such as business risks or commercial risks. Medical device risk analysis is concerned only with risks as defined in Article 2 MDR.
What role does Risk Management play within the MDR?
Risk Management is a central component of MDR Compliance. Risk Management is a direct component of Clinical Evaluation and a benefit-risk analysis is one of the required technical documents specified in Annex II MDR.
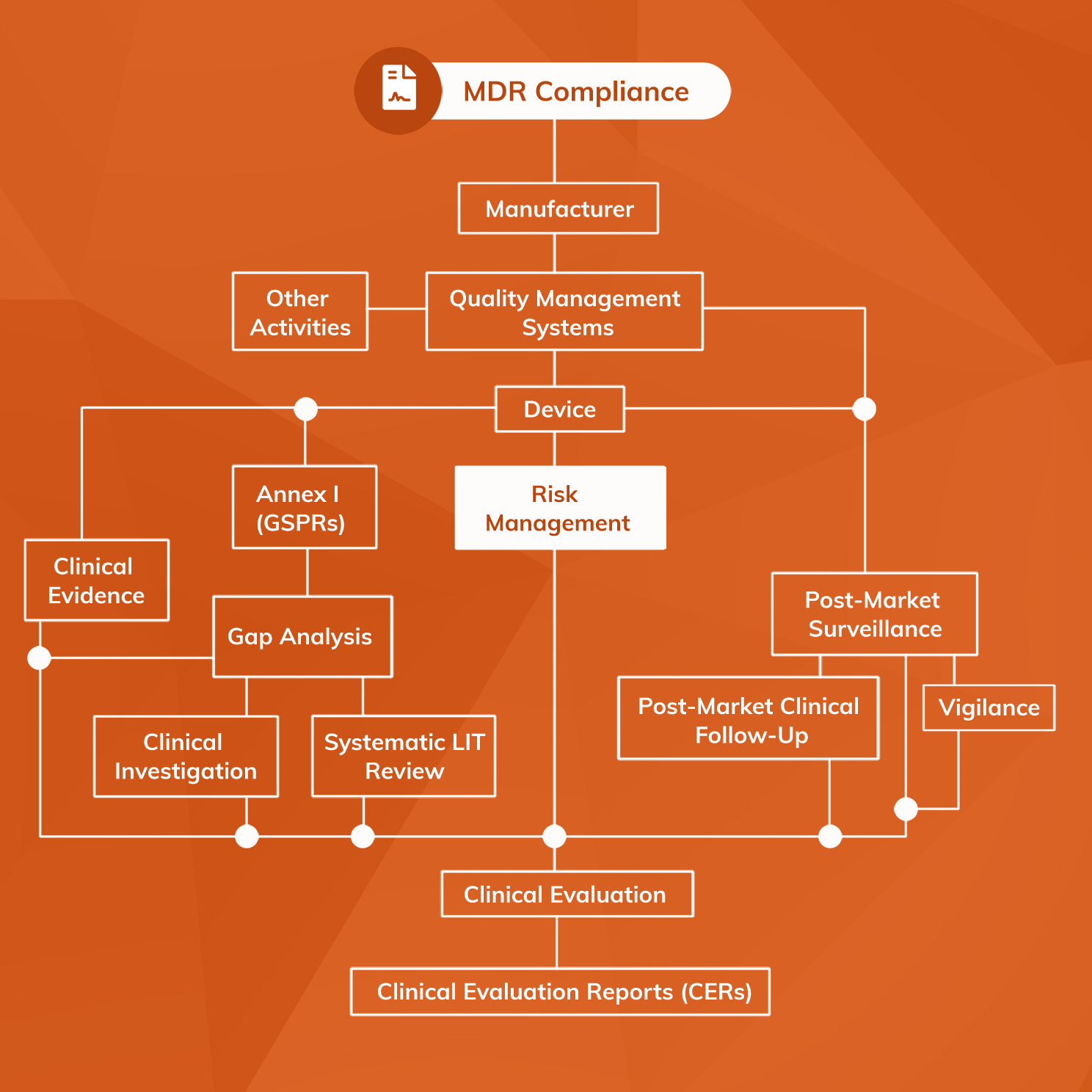
Article 10 MDR requires all manufacturers to establish, document, implement and maintain a system for Risk Management. Annex I provides greater detail about requirements for Risk Management stating that it shall be a continuous iterative process that is conducted throughout the entire lifecycle of a device.
Annex I states that manufacturers must:
- establish and document a Risk Management plan for each device
- identify and analyse the known and foreseeable hazards associated with each device
- estimate and evaluate the risks associated with, and occurring during, the intended use of the device and those resulting from any reasonably foreseeable misuse of the product
- eliminate or control identified risks
- evaluate the impact benefit-risk ratio and overall risk acceptability of any information arising from the production phase of the device and, in particular, from the post-market surveillance system
- if necessary, implement suitable changes to risk control measures
Annex I also requires that devices are designed to be able to withstand stresses, strains, temperature fluctuations, conditions of storage and transport, and environmental conditions to which they can be expected to be subject. Risk analysis therefore becomes a component of product design and must be documented from the initial product realisation phase onwards.
How to plan and develop a Risk Management strategy for medical devices
A compliant Risk Management strategy will involve the following stages (and associated documents):
- Overall Policy for Risk Acceptability
- Risk Management Plan
- Reasonably foreseeable misuse
- Characteristics related to safety
- Preliminary Hazard Analysis (PHA)
- Risk estimation & evaluation
- Risk Controls
- Overall Residual Risk
- Risk Management Review
- Post-Production Information
Planning a Risk Management strategy will require a combination of technical, regulatory and clinical knowledge. Detailed product knowledge and an understanding of the clinical context to which it will be applied will allow an initial risk matrix to be developed. The plan must:
- outline any assumptions made and provide justification for them
- detail strategies for confirming or refuting assumptions
- contain a plan for accurately determining frequency and severity of identified risks
- detail a plan for collating information about new or emerging product risks
- outline methods for determining risk acceptability
- detail a risk mitigation and risk elimination plan
- outline roles, responsibilities and reporting lines for members within the organisation whose activities may have a bearing on Risk Management
Risk Management documentation will form a component of the technical documents (Annex II MDR) that will be submitted as a component of the device conformity assessment process. Alongside the substantive Risk Management files it is necessary to document procedures for updating, maintaining, archiving and retrieving Risk Management documents.
Implementing a Risk Management strategy includes ensuring that activities documented with the Risk Management plan are undertaken in the correct manner. Risk Management activities interface with those conducted in running Vigilance systems, Post-Market Surveillance (PMS), and Clinical Evaluation, and so Risk Management activities are inherently cross-organisational.
Maintaining and updating a Risk Management strategy requires scheduled review and appraisal sessions to analyse system suitability. The clinical evaluation cycle offers an opportunity to assimilate Risk Management data collected and to re-perform a benefit-risk analysis of the device. Any updates or changes to the process must be reflected in documentation and disseminated across the organisation to ensure the changes are implemented.
What is ISO 14971?
ISO 14971:2019 - “Application of Risk Management to medical devices” is the most up-to-date version of the ISO 14971 standard. It closely reflects changes to Risk Management imposed by the MDR, but there are some differences that require consideration.
As with all internationally-recognised ISO standards relating to medical devices, ISO 14971 is regarded as a harmonised standard, meaning that compliance with the ISO standard will generally lead to a rebuttable presumption of conformity with most aspects of the MDR relating to Risk Management. However, whereas the ISO standard calls for reduction of all borderline and unacceptable risks, the MDR additionally requires reduction, where possible, of risks that are already acceptable.
ISO 14971:2019 outlines a process for Risk Management and extends its coverage to software as a medical device and in-vitro diagnostic medical devices. It can be applied to all phases of a product’s life cycle.


