Software as a Medical Device (SaMD)
A guide to achieving MDR compliance for medical device software
What is software as a medical device?
The term ‘Software as a Medical Device (SaMD)’ applies to software products that meet the definition of ‘Medical Device’ in EU MDR Article 2. SaMD is an increasingly important sector of the medical device market, as rapid advances are leading to the potential to improve care across the spectrum of healthcare. However, applying the MDR to a software product is not without challenges.
Regulating software as a medical device is not new to the MDR; in fact, software of some form has been regulated as a medical device since the introduction of the Medical Device Directive in 1993.
Medical device software is any software product that meets the following criteria:
- it is intended by the manufacturer to be used for a medical purpose
- it meets the MDR Article 2 definition of “medical device”.
The growing range and sophistication of medical software means that this aspect of medical device regulation is becoming increasingly important. The ever-expanding selection of medical apps, AI diagnosis and diagnostic-assist technology, medical imaging analysis software, decision support tools and other medical software solutions mean that software poses a different set of regulatory challenges to physical products when gaining certification under MDR.
When is a software product a medical device?
Depending on the nature of the product, determining whether a software product is a medical device can pose difficulties. Fortunately, the guideline MDCG 2019-11 sets out a useful scheme for qualifying and classifying software that is easy to follow and logical. Key considerations are the intended purpose of the product as stated by the manufacturer, and any specific claims made about product functionality.”
Article 2 of the MDR contains a wide definition of “medical device” which extends to any software that is intended for diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease, and/or diagnosis or monitoring of an injury or disability.
The nature of software, however, is that even with such a broad definition it can still be unclear whether the EU MDR will apply. Some software products, for example, have a functionality that enables healthcare activity but does not have a stated medical intended purpose (for example, software that co-ordinates shift patterns of healthcare workers, or inventory-management software). Other software may make healthcare-related information more accessible while claiming not to alter the information in any way (such as an app that shows a trend of blood sugar readings over time).
The following additional considerations may be useful in further understanding whether a software product will fall under the MDR.
1. The software must have a medical purpose on its own
This applies whether the software is a stand-alone product, or whether it is integrated into a hardware device. For example, the software that drives decisions on automated defibrillators is embedded in a hardware device; however, it would itself be a medical device if it was extracted from the defibrillator
2. The software must add medical value
Simple “archive and retrieval” software that, in effect, replicates the action of a paper recording of healthcare data is not likely to be classed as software as a medical device. However, if the software highlights data in any way (especially if it applies an analysis to trends or draws attention to potentially significant findings) then this would be classed as “adding medical value”, making it likely that the MDR would apply.
3. Software incorporating a “call to action”
Any “call to action” initiated by the software and related to any of the aspects covered by MDR Article 2 will likely cause the product to be software as a medical device. For example, an app that advises the patient to visit their doctor based on a reading or data entry is likely to be classed as software as a medical device.
4. Do virtually-identical systems operate outside the medical device sector?
An increasingly-relevant example are systems that enable remote medical consultation. If the system is simply a communication aid (such as video-conferencing software that could equally be used, unaltered, for non-medical purposes) then it is unlikely to be software as a medical device. Systems that enable remote consultation but also assist the provision of care in any way (for example, by highlighting important information) are likely to fall under the MDR.
What regulatory systems need to be in place for software as a medical device under the MDR?
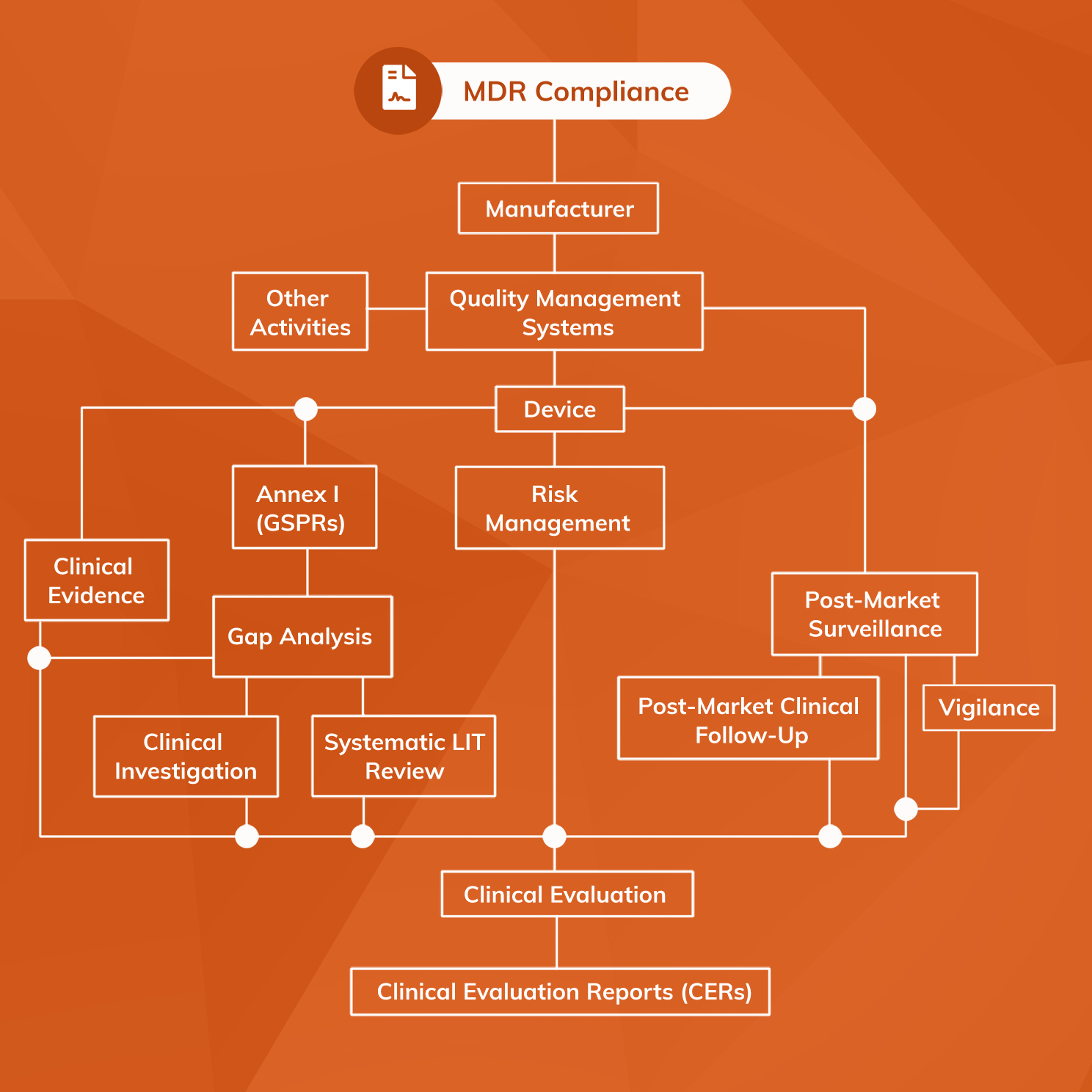
Overall, software as a medical device must meet the same range of regulatory requirements as physical medical devices before application of a CE-mark. However, the application and handling of MDR requirements are necessarily different for SaMD. Software products inherently evolve far more rapidly than physical products, and the technical complexity of generative AI poses challenges for ensuring Notified Bodies can comprehend material within the technical file. Verification must demonstrate suitable software security and stability, while validation activities must show that the SaMD’s outputs and the target clinical condition are scientifically associated.
SaMD is also complicated by the fact that it can fall into any one of the four available risk classes (Class I, IIa, IIb or III). The key to understanding risk classification of software as a medical device is MDR Annex VIII Rule 11, which sets out a classification system that can be applied to any medical software product. While there is scope for software being in any of the four risk classes, by virtue of Rule 11 most software as a medical device will fall into Class IIa.
Manufacturers of all devices (including software) apart from Class I devices must gain approval from a Notified Body before applying a CE-mark.
Specific regulatory processes that must be in place for software as a medical device include:
- Risk Management system
- Quality Management System (QMS)
- Post-Market Surveillance (PMS) system, including PMCF and Vigilance components
- the performance of a Clinical Evaluation and compilation of a Clinical Evaluation Report (CER)
- the production of a full dossier of Technical Documents according to MDR Annex II and III.
Overall, software as a medical device requires a specialised regulatory approach that combines technical comprehension of the software with the ability to properly apply MDR requirements to SaMD products. For this reason, Mantra Systems offers a consulting team that specialises in preparation of SaMD technical files, offering you peace of mind throughout your approval journey.


