Post-Market Surveillance (PMS) under the EU MDR
A complete guide to designing & implementing PMS systems for medical devices
What is Post-Market Surveillance (PMS)?
Video 1: A 3 minute summary of Post-Market Surveillance (PMS) under the EU MDR
Post-Market Surveillance (PMS) is defined as a structured process of monitoring the safety and performance of a medical device following its release onto the market.
Effective Post-Market Surveillance requires a combination of processes and systems that work together to develop a robust ‘picture’ of how a medical device is performing in real-world use following its commercial release.
The MDR builds upon previous requirements for PMS systems in medical devices. It requires that a Post-Market Surveillance framework is in place for all medical devices, using a design that is proportionate to risk class and appropriate for device type.
What are the MDR requirements for Post-Market Surveillance?
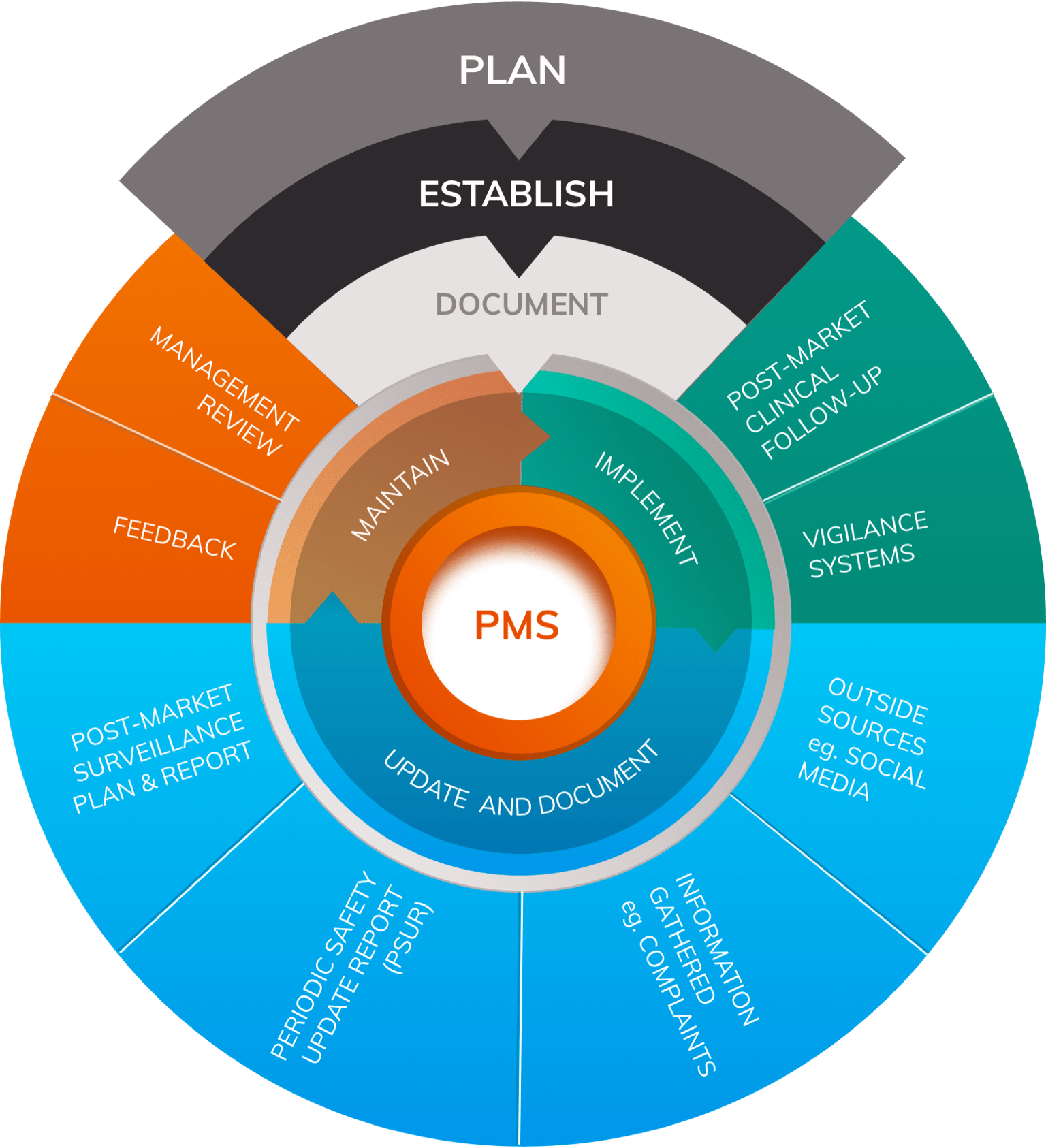
MDR Article 83 outlines the requirements for Post-Market Surveillance systems under the new regulatory framework. Every manufacturer is required to plan, establish, document, implement, maintain and update a PMS system for every medical device. The system must be designed and documented in a PMS plan (detailed below) and then constantly updated according to a structured assessment of how well it is working and ongoing analysis of the data it has yielded.
MDR Article 83 requires that data generated by Post-Market Surveillance systems is used in specific ways; in particular it should be used to:
- Update the benefit-risk profile of the device through contributing data about the frequency and severity of device-related harms
- Feed improvements to Risk Management processes
- Update device design, Instructions For Use (IFU) and device labels to optimise device safety and performance during use
- Form a core component of updating the Clinical Evaluation of the device
- To identify needs for Preventive And Corrective Actions (PACAs) or Field Safety Corrective Actions (FSCAs)
How does an effective Post-Market Surveillance system monitor medical device safety?
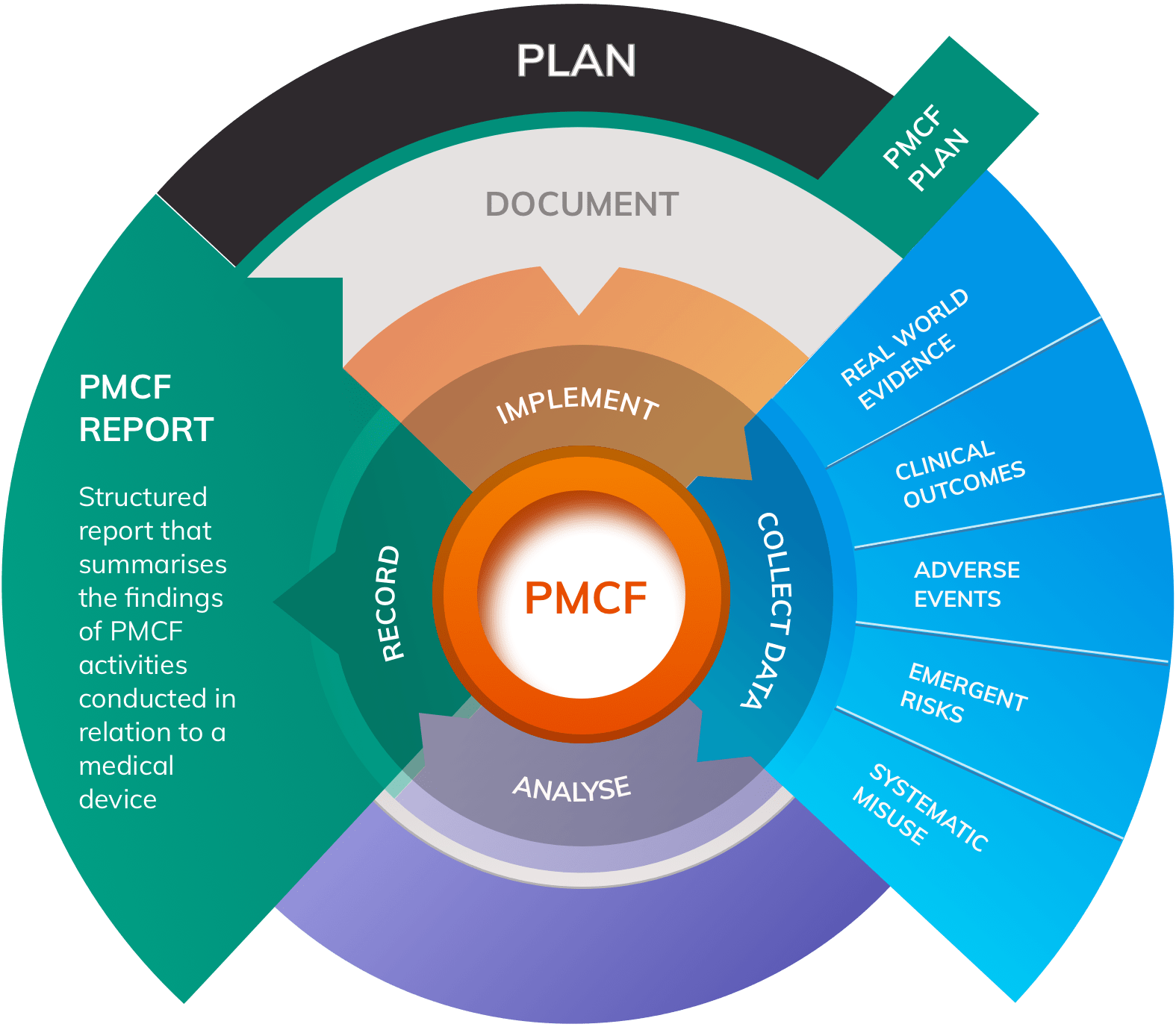
A well-designed PMS system will monitor the safety and performance of a medical device through two complementary domains:
- Post-Market Clinical Follow-up (PMCF) that involves the design and conduct of clinical studies to proactively and continually assess device safety and performance
- Vigilance systems that monitor and respond to complaints, adverse events, media reports, serious incidents and FSCAs
Vigilance systems can be thought of as an “ear to the ground” for any potential safety or performance concerns relating to the device. Although rare in practice, it is at least theoretically possible for vigilance systems to return no data. On the other hand, PMCF involves the continuous collection of clinical data throughout the entire clinical lifetime of the device, generating Real World Evidence (RWE) on safety and performance. In contrast to vigilance, Post-Market Clinical Follow-up will always return some data by virtue of design.
Post-Market Surveillance systems that incorporate both arms of data collection are more robust and will more closely align with MDR requirements.
What documents must be produced in relation to MDR PMS systems?
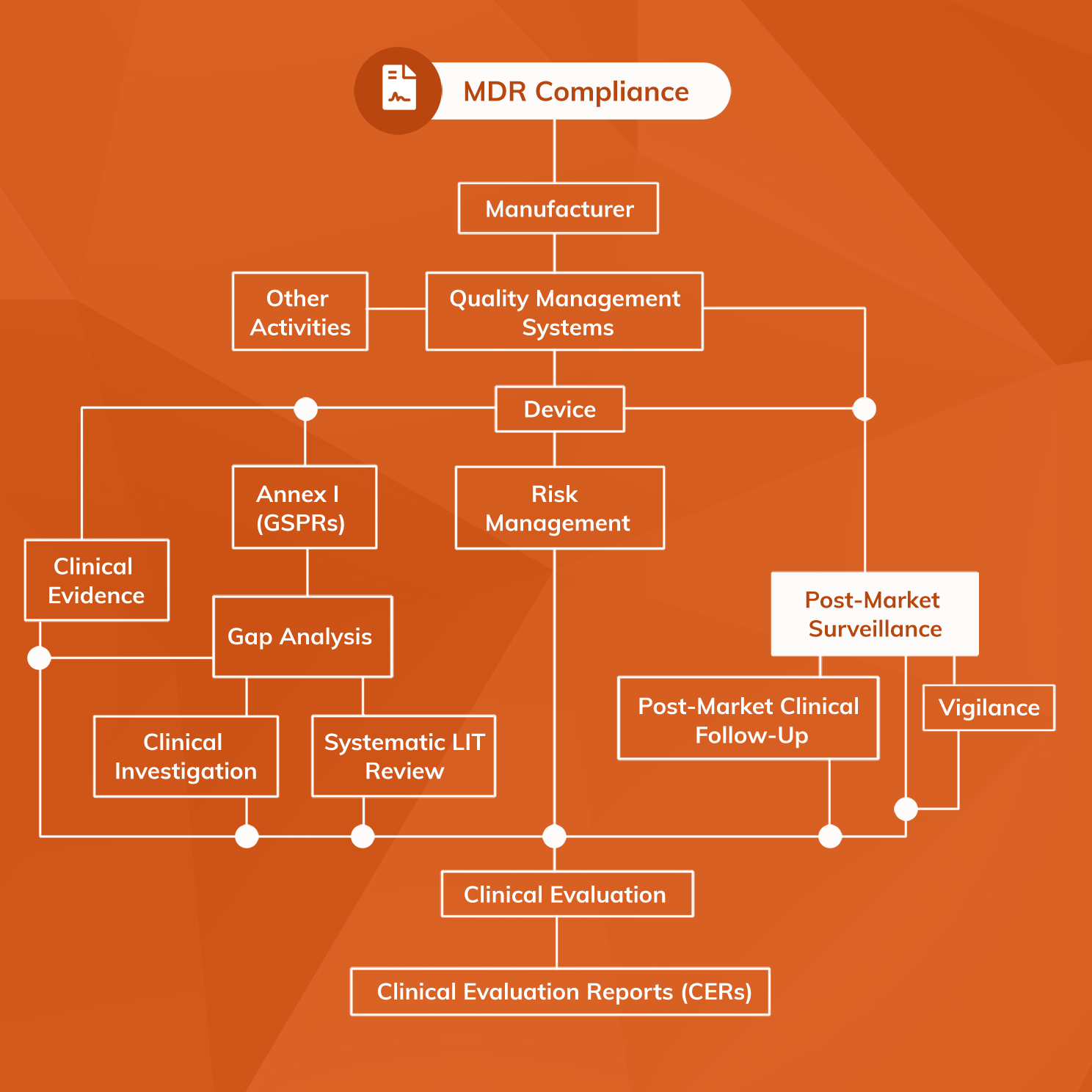
MDR Annex III provides useful details about what technical documents must be produced in relation to Post-Market Surveillance.
All MDR PMS systems require a PMS Plan that describes in detail how the system will collect and analyse safety and performance data. A well-written PMS Plan will account for using data from a wide range of different sources including:
- Serious incidents
- Field Safety Corrective Actions (FSCAs)
- Complaints
- Databases and/or registries
- Feedback and complaints
MDR Annex III outlines rules for constructing the PMS Plan. As a minimum the PMS plan should contain the following:
- A documented process to collect information from the identified data sources
- A description of methods that will be used to assess and interpret collected data
- An explanation of the rationale used during risk analysis
- Detail of methods for collecting and investigating complaints and analysing trends
- Procedures for communicating with competent authorities and Notified Bodies
- Methods for tracing devices following product complaints or adverse events
The output of PMS activities must be collated in a PMS output report. The format of this report varies according to the device’s risk class:
- For Class I devices a comparatively simple PMS Report is required that simply summarises outcomes from PMS activities relating to the device.
- For all other device classes a more detailed Periodic Safety Update Report (PSUR) must be produced with a periodicity that varies according to risk class (at least annually for Class III).
What are MedDev 2.12/1 Rev 8 and MedDev 2.12/2 rev 2? How do they relate to PMS under the MDR?
The European Commission produces a range of guidance documents, known as MedDev guidelines, in relation to medical device regulation. MedDev guidelines are not legally binding but are a very useful source of information when developing PMS systems and other aspects of medical device regulation.
MedDev 2.12/1 Rev 8 provides guidance on medical device vigilance systems, while MedDev 2.12/2 rev 2 details guidelines for PMCF. Although neither has been updated to reflect changes introduced by the MDR, they are still useful documents that are well worth reviewing.
Mantra Systems offers tailored PMS consulting services on a flexible basis, keeping you compliant and your devices on the market. Speak with a member of our team in complete confidence today.


