EU MDR Compliance Guide
Work through our free guide & use our online resources to build your Medical Device Regulation strategy
Looking for expert consulting support on your regulatory strategy? Contact us today for a free & confidential discussion about your options.
What is EU MDR Compliance?
EU MDR compliance is accomplished by demonstrating conformity with all relevant aspects of the Medical Device Regulation (EU) 2017/745. Manufacturers will need to demonstrate compliance in order to gain regulatory approval of their medical devices.
Video 1: A 3 minute summary of EU MDR compliance
Medical devices must be designed, manufactured, distributed and tracked according to EU MDR requirements, and manufacturers must develop a suite of compliant regulatory systems, processes and documents to continually monitor the safety and performance of their products.
EU MDR compliance begins with a detailed understanding of the regulation and the obligations it imposes upon manufacturers. In this comprehensive free guide, written by our experienced experts, we outline essential requirements in detail and cover all aspects of the medical device regulatory framework. If you have any questions about the information in this guide please talk with a member of our team.
What are the main components of EU MDR Compliance?
Understanding the EU MDR
Gain clarity & context on overall EU MDR requirements for your entire regulatory strategy
At the most elementary level, EU MDR compliance can be considered in three principal domains:
- The medical device
- Clinical evidence, suitably generated, appraised and analysed
- Regulatory systems, processes and documents
1. Medical devices
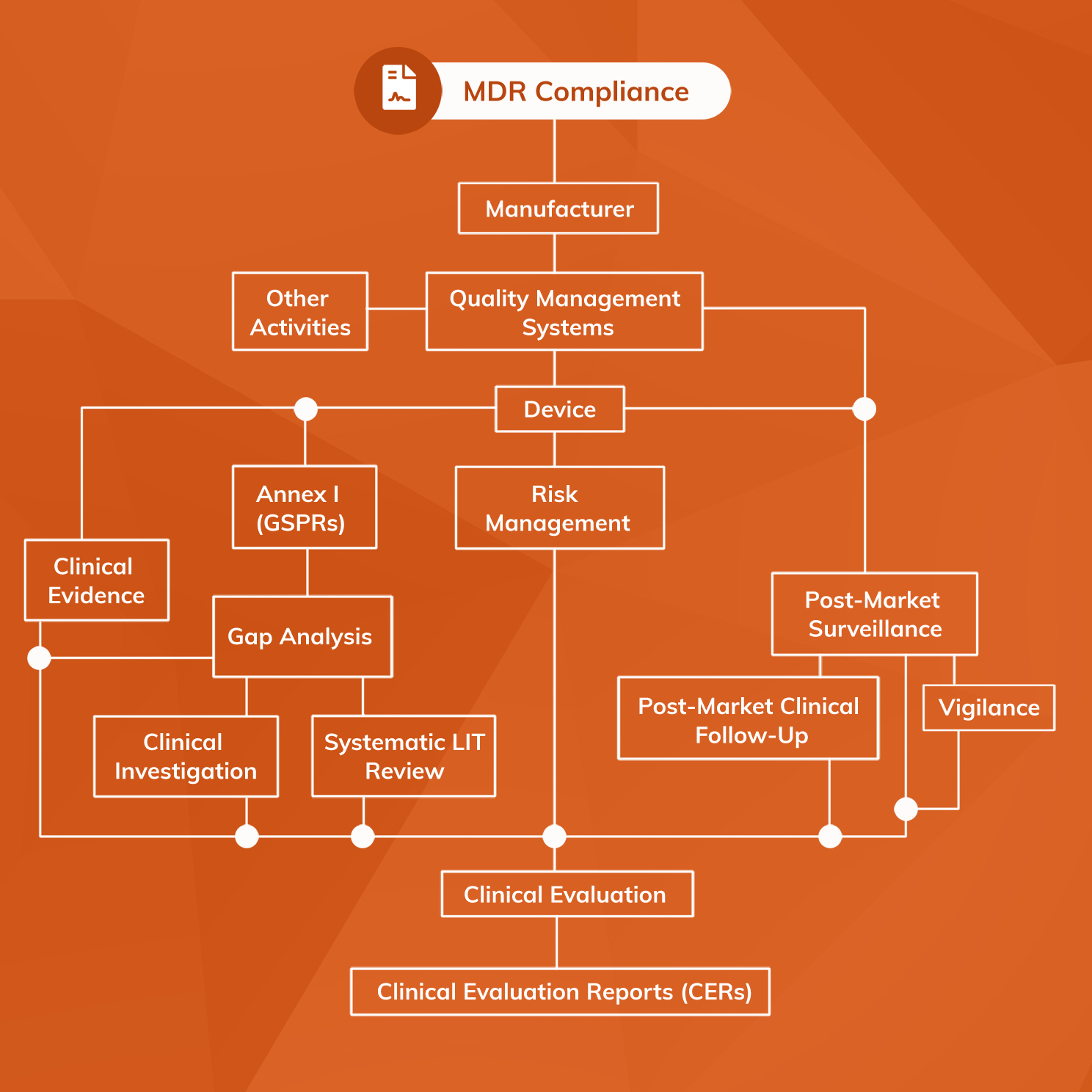
Medical devices must be suitable for their intended purpose, demonstrate a favourable benefit-risk profile and comply with all relevant MDR Annex I General Safety and Performance Requirements (GSPRs). Devices must be developed within an appropriate Quality Management System (QMS), with most manufacturers applying ISO 13485:2016 as a harmonised standard to ensure QMS suitability.
2. Clinical evidence
Clinical evidence must be produced and identified in relation to all medical devices under the EU MDR. Conclusions about suitability of the device to its intended purpose, GSPR conformity, and benefit-risk profile must be supported by clinical evidence that has been appropriately appraised and analysed. Clinical evidence includes that generated and held by the manufacturer, as well as data published independently in journals and other sources.
3. Regulatory systems, processes and documents
Regulatory systems, processes and documents required for EU MDR compliance are more extensive than those necessary under the out-going Medical Device Directive (MDD). Those required for MDR Compliance include the following:
- Quality Management System (QMS) covering the activities of the entire organisation
- Post-Market Surveillance (PMS) system monitoring safety and performance of each device following release onto the market
- Post-Market Clinical Follow-up (PMCF) and Vigilance system for each device
- Risk Management system to the standard in ISO 14971:2019 or equivalent
- Clinical Evaluation process for every medical device, documented in a correctly-structured Clinical Evaluation Report (CER) produced at appropriate intervals
- A dossier of Technical Documents according to requirements in Annex II and Annex III
- A Person Responsible for Regulatory Compliance (PRRC) either within the organisation or permanently at its disposal
Mastering the MDR
An easy-to-digest summary of the MDR, highlighting the areas most relevant to medical device manufacturers
Rules for assessing compliance differ according to the risk class of the device. For Class IIa, IIb and III devices a Notified Body will be required to perform an assessment of the device, technical files, and regulatory systems to determine whether MDR Compliance has been achieved. By contrast, most Class I devices do not normally require involvement of a Notified Body but must still be supported by MDR-compliant regulatory files, systems and processes. Class I devices that are sterile or that have a measuring function must seek approval from a Notified Body.
Successful compliance is demonstrated by the application of a CE-mark to the device. Class I manufacturers (except for devices that are sterile or have a measuring function) may self-apply a CE-mark after producing a declaration of conformity. For all other classes of medical device, a CE-mark may only be affixed once a Notified Body has issued a certificate of conformity following a regulatory review according to rules in Chapter IV of the MDR.
Please note that the MDR compliance process is essentially the same for physical medical devices and any digital products that meet the definition of Software as a Medical Device (SaMD).
How to achieve EU MDR Compliance
Video 2: How to build an EU MDR compliance strategy for your medical devices
The most effective route to achieving EU MDR compliance has two distinct phases:
- Develop a deep understanding of the MDR and associated guidance documents
- Apply that understanding to build a framework that complies with MDR requirements
Developing a deep understanding of the regulatory framework is a crucial first step in achieving MDR compliance. Our range of free resources and articles can support your team in developing the necessary level of MDR knowledge.
1. Understand the EU MDR in detail
The text of the MDR contains the basis for the rules that regulators will apply when assessing every medical device submission.
The Medical Device Regulation MDR 2017/745, commonly known as the MDR, is an extensive piece of legislation that represents the largest overhaul of medical device regulation in decades. Now scheduled for full implementation in 2021, the EU MDR replaced the Medical Device Directive MDD 93/42/EC and introduced a wide range of changes in the way medical devices are regulated in Europe.
The MDR describes a detailed regulatory environment that outlines rules and regulations for all aspects of developing, marketing and monitoring the performance of medical devices. It is therefore essential that medical device manufacturers develop a detailed understanding of the MDR or risk falling short of requirements.
The MDR introduces a range of changes that place a higher burden on manufacturers in many areas, including:
- A wider definition of the term “medical device” — The scope of the MDR is wider than that of MDD meaning that some product manufacturers, such as those that make medical software applications or nano materials, are subject to medical device legislation for the first time.
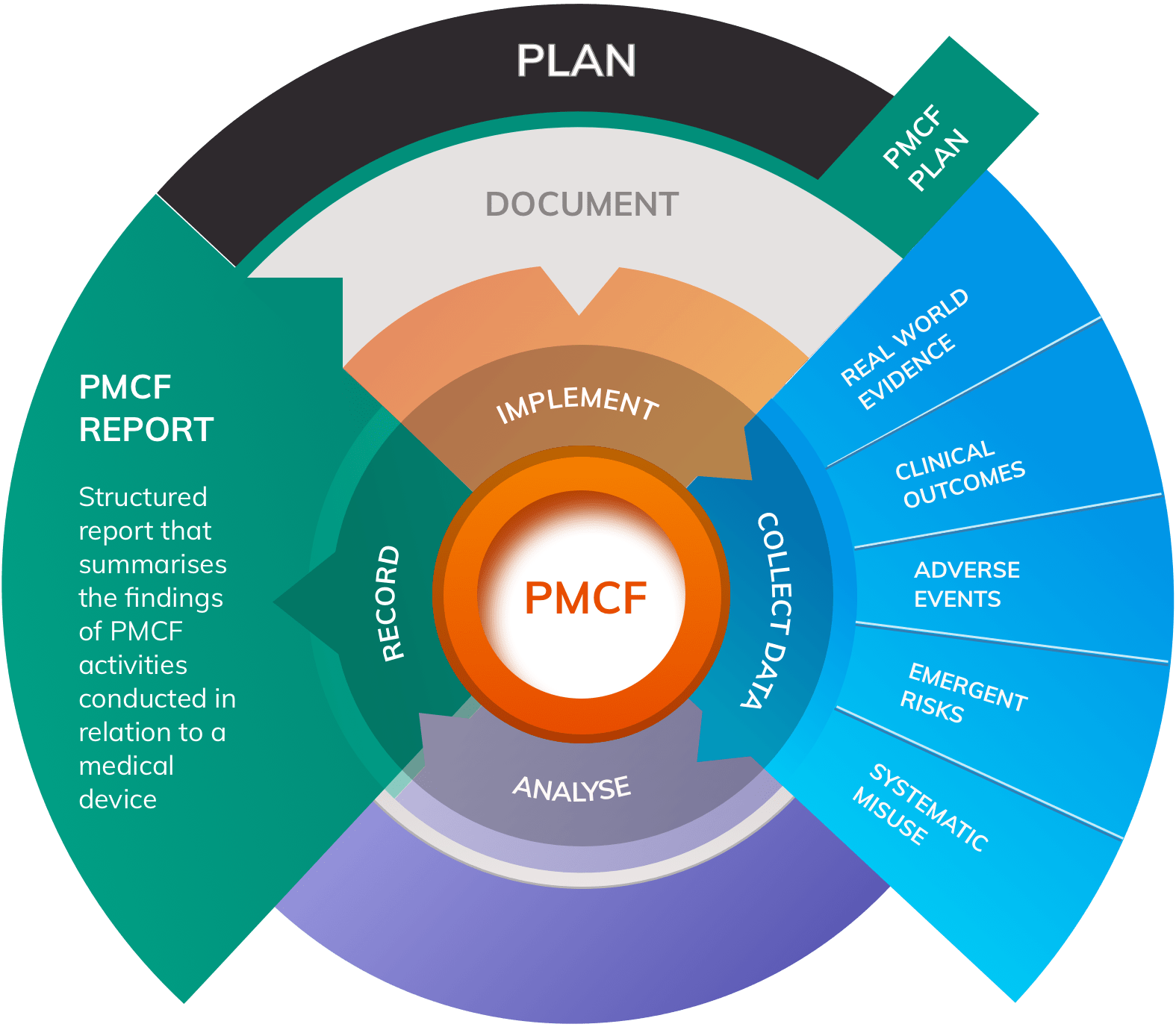
- Tougher requirements for clinical evidence — All available clinical evidence, whether favourable or unfavourable, must be identified in relation to the subject device and any comparable alternatives. Evidence must be appraised and analysed according to a specified protocol. Alongside conventional clinical evidence, Real World Evidence (RWE) must be produced through PMCF systems that demonstrate safe and effective performance of the device in the hands of the ‘normal user’.
- Limitations on equivalence — Under MDD it was possible for manufacturers to claim that their device was ‘equivalent’ to a related device already approved for sale; they could then “piggy back” on the clinical evidence portfolio of that equivalent device without needing to produce their own. Under MDR, the ability to do this has been significantly constrained.
It is also important to keep updated with harmonised standards such as ISO standards, compliance with which will lead to a presumption of compliance with the MDR in that area. In addition, the various MedDev guidelines provide guidance in structuring and performing a range of MDR Compliance activities.
2. Apply your understanding to build an MDR-compliant framework
A thorough understanding of the MDR allows you to apply this knowledge, along with guidance from harmonised standards and MedDev guidelines, to begin constructing MDR-compliant systems and processes. Many regulatory processes that are required for MDR Compliance follow the same cyclical structure for their implementation and maintenance which should be reflected in their design. This system is usually:
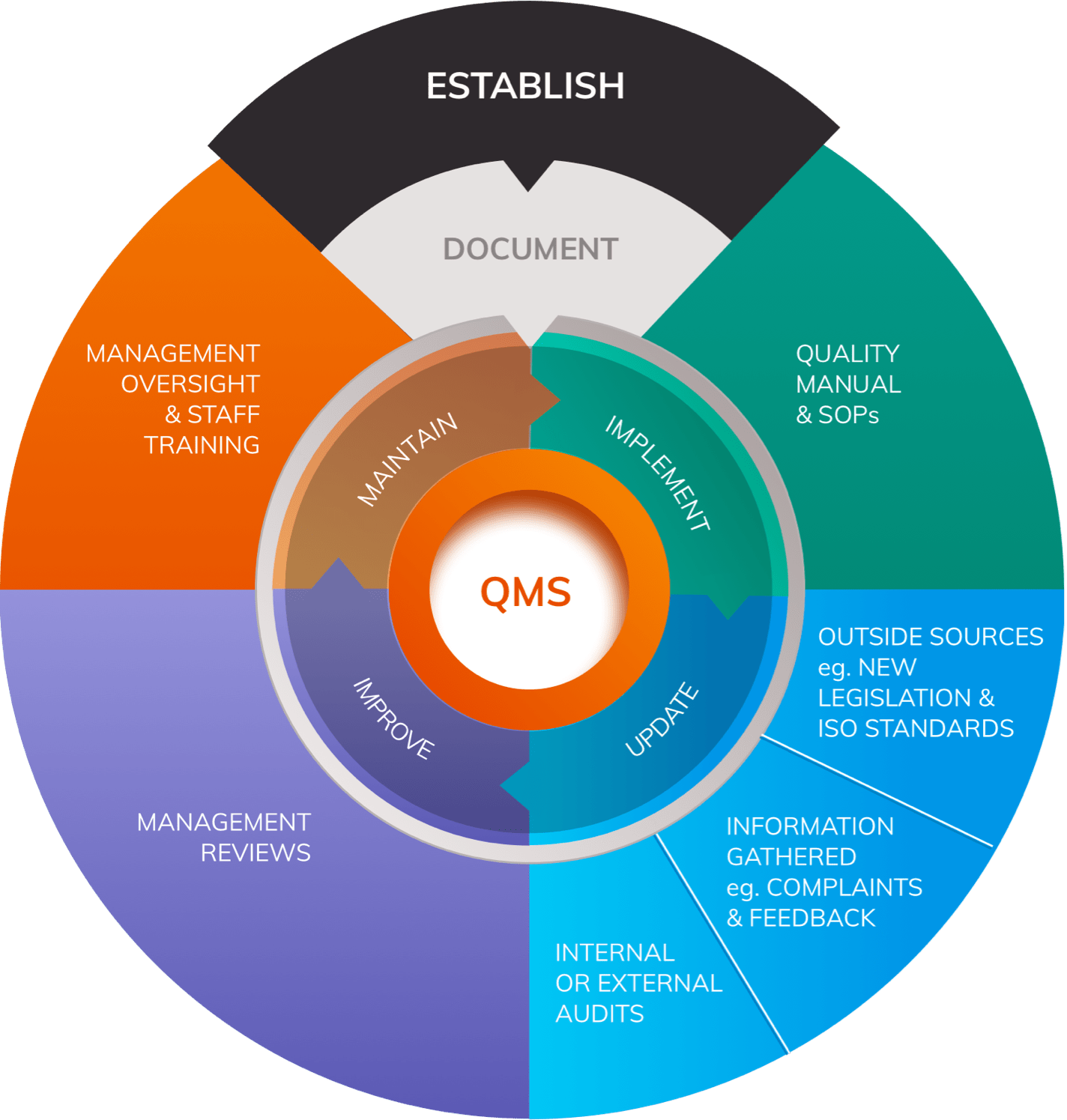
- Establish
- Document
- Implement
- Update
- Improve
- Maintain
Applying this process to Quality Management Systems, PMS Systems, Risk Management processes and Clinical Evaluation will help ensure that information gathered during the “Implement” phase is worked back into the design of the systems and responded to appropriately.
Even with a thorough understanding of the MDR, building compliant systems can be challenging. For example, demonstrating conformity to the GSPRs, developing PMCF procedures within a PMS system, and performing Clinical Evaluation may all involve the design, documentation, implementation, conduct and interpretation of clinical investigations. Performing and interpreting clinical evidence requires a degree of medical insight that many medical device companies do not have available in-house.
Clinical evidence in the EU MDR
Clinical Evidence Generation Systems
PMCF Studies, Surveys & Plans for all classes of medical device.
Clinical evidence has assumed an even greater level of importance in demonstrating device safety and performance under the MDR than it did under the MDD. MDR Compliance will require a comprehensive clinical evidence portfolio that demonstrates:
- Safety and efficacy for every individual indication in normal use
- Conformity to the Annex I General Safety and Performance Requirements
- Acceptability of benefit-risk ratio
- Acceptable levels of side-effects and adverse events
- Safe and effective real-world use
- Adequate responses to serious incidents
Meeting these requirements will necessitate:
- Performing literature searches, critiquing sources, and performing a scientific analysis and report of findings
- Designing Clinical Investigations in line with Good Medical Practice, Annex XV MDR, and the GDPR
- Applying different types of Clinical Investigation to different purposes
- Understanding how to work with clinical sites to enhance data collection compliance
- Ensuring requirements for patient consent and ethical approval are met
- Implementing well-structured Vigilance systems and applying a medical interpretation to the results
Furthermore, regulatory staff need to have a good working relationship with marketing departments and sales staff, since all marketing and promotional claims made in any marketing material must be backed by appraised clinical evidence.
It is not enough simply to produce clinical evidence; the methods by which such evidence is generated must also be compliant with requirements. Clinical Investigations must minimise the potential for bias, be adequately powered, represent the population normally subject to the device, and employ appropriate methods of statistical analysis in interpreting and applying results.
Methods must be outlined in a range of technical documents that will be subject to regulatory scrutiny. Annex XV MDR determines that necessary documentation includes a Clinical Investigation Plan and Investigator’s Brochure for each Clinical Investigation, as well as any other information deemed necessary to ensure proper conduct of the study and scientific validity of results.
It should be clear that designing Clinical Investigations intended to be part of any MDR compliance strategy is a specialist task that requires extensive clinical, regulatory, legal and medical device industry experience. Many companies will fail to produce clinical evidence of the standard required because they do not have access to such expertise in-house, threatening the regulatory approval status of their products. In particular, generating clinical evidence draws upon a deep understanding of clinical investigation design and medical insight.
How is the race for MDR Compliance affecting the industry today?
MDReady Starter Guide
Begin your EU MDR compliance journey with a free step-by-step guide
Although the MDR is not yet in full force, many manufacturers from all sectors of the industry are facing challenges in meeting the requirements for EU MDR compliance. Meeting the requirements of the MDR in time will require many manufacturers to implement new policies and strategies well ahead of the deadline.
- Meeting the requirements for MDR compliance by the 2021 deadline will require forward planning in order to ensure that evidence and regulatory systems are ready for submission in time.
- Long-held claims of equivalence may no longer be valid meaning that, in many cases, new evidence portfolios will need to be produced from scratch.
- The need to evidence every indication is placing huge demands on evidence-generation systems that will need to be expanded or re-designed in order to meet needs.
It is estimated that nearly 80% of European medical device companies do not yet have a regulatory strategy that would be sufficient for MDR compliance. This highlights both the low level of understanding and the low level of preparedness towards regulatory compliance within the industry. Many manufacturers, without a change in approach, run a real risk of regulatory approval being removed for their products after the 2021 deadline.
Why has the Medical Device Directive (MDD) been replaced?
Although generally robust and relatively permissive of innovation, the MDD was considered to have numerous flaws in enforcing medical device safety, reliability and general product quality, exposed by notorious incidents such as the metal-on-metal hip scandal. Legislators were compelled to implement reform.
The new MDR attempts to reduce the likelihood of future incidents of this nature by enhancing requirements for proving safety and effectiveness of all medical devices, regardless of how long the product has been on the market.
The five most important changes you will need to know about
- Broader definition of “Medical Device”. Under the MDR, diagnostic software, software applications, nano materials and some products that specifically claim not to be medical devices are amongst those that will be caught by the Article 2 MDR definition of what is a medical device.
- Reduced ability to claim Equivalence. Claiming equivalence to another medical device already approved for sale will now require full unfettered access to the technical files of that medical device - something that would appear unlikely if the equivalent device is made by a competitor.
- A change to risk classification of some categories of device. Generally, where any changes have been introduced they have resulted in a higher risk classification, increasing the regulatory burden on manufacturers.
- Reduced Notified Body capacity. As of July 2024, only 49 Notified Bodies have gained approval to perform conformity assessments under the MDR, forming a bottle-neck and making it even more important that manufacturers develop an MDR compliance strategy well ahead of the May 2021 deadline.
- An increased emphasis on clinical evidence and PMCF. More than ever before, device safety and performance, benefit-risk analysis, Clinical Evaluation and Post-Market Surveillance will require the production of high-quality clinical data including robust Real World Evidence (RWE). The increased role of RWE in demonstrating safety and efficacy of a device in normal use runs in parallel with the MDR’s emphasis on the importance of PMCF.