PMCF has undergone significant changes with the introduction of the EU MDR, with increased focus and regulatory scrutiny of PMCF systems. As a result of these changes, many manufacturers of established products have been left wondering whether they need to develop PMCF systems for their medical devices.
In this article, we outline why even long-standing legacy medical devices may need a PMCF system under the EU MDR. We also set out a range of reasons why PMCF could be avoided if a device meets a set range of criteria.
For advice on developing PMCF systems for your devices, contact our experienced team for a free, no-obligation discussion.
“I have a well-established legacy device - surely I don’t need a PMCF system?”
Legacy devices with a long clinical history would seem unlikely candidates for needing a PMCF system. After all, it could justifiably be argued that a long, successful clinical history stands as plentiful evidence of a device’s safety and suitability for purpose.
If an established legacy device has been entirely consistent throughout its history without undergoing any substantial changes in indication, intended purpose or patient population, then it is indeed possible that an exemption to PMCF could be successfully argued. In fact, long-standing devices unchanged through their commercial journey are the least likely of all devices to need a dedicated PMCF study.
Exceptions
There are exceptions, however, that could mean a legacy device would still need to produce PMCF data on an ongoing basis, no matter how well established:
- It is a high-risk device (high risk classification, vulnerable patient population, device incorporates dangerous substances)
- There have been changes to the device’s indications, target patient populations, IFU, labelling or packaging since original CE-marking
- Novel changes have been introduced to the design of the device
- Clinical data is very old and has not been updated for some time
- Risk Management and/or PMS activities have led to safety or performance questions relating to the device or to similar devices on the market
- A regulatory body has advised that a PMCF system needs to be in place
If any of these criteria apply to your device, the chances are that a PMCF system will need to be developed to ensure ongoing regulatory approval. Contact our team for personalised advice on how to design product-specific PMCF systems for legacy devices.
“My device is a low-risk Class I device”
The applicability of PMCF to Class I devices can be understood by considering the main reasons for performing PMCF on any device under the MDR.
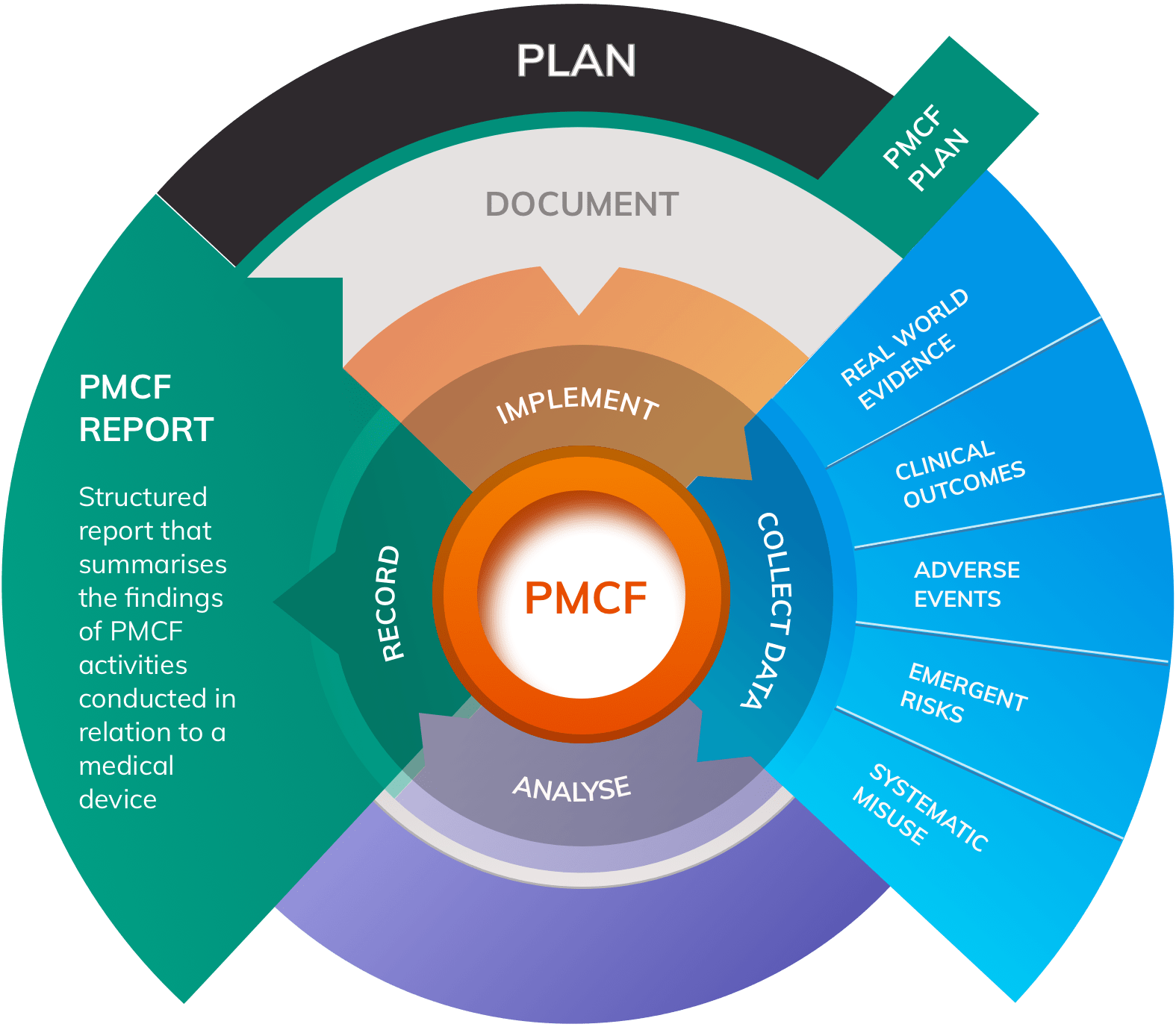
Principal objectives of PMCF include:
- Collecting data on new or existing side effects
- Monitoring for adverse events relating to use of the device
- Monitoring for systematic misuse of the device in practice
- Detecting any emergent risks that have not been considered in the device’s Risk Management activities, and harvesting data on the frequency and severity of harms identified in existing Risk Management documentation
- Collecting ongoing information about the safety and performance of the device in normal use
Even a Class I device would normally be expected to exhibit side effects and risks, and all such devices must have a risk management system as part of their regulatory framework. Unless very well established as a legacy device, in many cases it will therefore be necessary for Class I device manufacturers to ensure that PMCF systems are in place for their devices.
This considered, it is certainly worth making the point that not all PMCF systems are equal - and nor should they be. While a Class III implantable device may require a sophisticated medical device product registry for their PMCF activities, a simple Class I product could be served by a much less complex but a well-designed PMCF survey which would be far less expensive and easier to implement in practice.
Ensuring suitability of PMCF system design for purpose is a core feature of our PMCF design service. Save money, time and resource by working with our team of experts when producing PMCF systems for your devices.
“My device is supported by RCTs showing that it is safe and performs as intended”
Randomised Controlled Trials (RCTs) represent one of the highest levels of clinical evidence. Manufacturers of products supported by high-quality clinical evidence such as RCTs or systematic reviews may feel that a PMCF study producing “inferior” evidence is therefore an unnecessary waste of resources.
The reasons this argument may be unsuccessful is that PMCF data is not ‘inferior’ to RCT data; rather, it has a different function and generates a different type of evidence to level I studies like RCTs.
RCTs, by definition, involve randomisation of subjects into one of two or more groups. By defintion, therefore, RCTs are comparative in nature. Furthermore, part of the power of RCTs comes from making the different groups as comparable as possible, which in turn calls for stringent inclusion and exclusion criteria for use when recruiting subjects into the study. The outcome of RCTs is accordingly ‘clean’ data that enables a valid comparison between groups.
PMCF data is not intended to be comparative; rather, it should focus only on performance of the device in question. Furthermore, PMCF data should not be ‘clean’ in the sense of subject recruitment being driven by strict inclusion/exclusion criteria. Ideally, a PMCF study should collect real world evidence on the safety and performance of a device in actual clinical use, outside the inclusion/exclusion constraints of a formal RCT study. To take an obvious example: a RCT would (by definition) be unable to collect data on systematic misuse of a device, whereas a well-designed PMCF study should include provision for collecting (but not encouraging) instances of systematic misuse as a component of its design. Finally, RCTs collect high-quality data over a limited period of time while PMCF should collect targeted data throughout the entire clinical lifetime of a device, fulfilling an entirely different clinical purpose.
Overall, the existence of robust RCTs does not automatically mean that PMCF will not be required. If in doubt, a free discussion with a member of our team may help you decide whether or not PMCF will be needed for your device.
Summary
Despite the increased focus and importance of PMCF under the EU MDR, there do remain legitimate justifications for not implementing PMCF in certain circumstances. In practice, though, these occasions are limited to well-established low-risk devices that are essentially unchanged since initial CE-marking and have demonstrated no alteration in risk profile since launch. Additionally, non-performance of PMCF would not normally be justified in the absence of any recent clinical data or where evidence fails to address all indications or target groups for the device.
In practice, it may often be advisable to focus on effective and device-appropriate PMCF design than to undertake convoluted justifications for non-performance. However, a reasoned consideration of the applicability of PMCF to your device is an important part of planning your regulatory strategy.
Good quality advice is key in making effective PMCF decisions. Our team are here to help with simple, straightforward and honest advice about PMCF and its applicability to your medical devices. Call us today for a free consultation.
This article is part of a series covering all aspects of PMCF under the MDR. Subscribe to our Newsletter to ensure you don’t miss any of this comprehensive series.