With the transition to the EU MDR, legacy devices with certificates issued under the old EU directives (Directive 93/42/EEC on medical devices, MDD, and Directive 90/385/EEC on active implantable medical devices, AIMDD) can stay on the market under the transitional provisions set out in Article 120(3) of the MDR, provided they do not undergo any significant changes.
In this article, we explain the concept of a ‘significant change’ and, using examples, help you understand whether you can still take advantage of the transitional arrangements in MDR Article 120(3).
Transition periods
The transition periods in Article 120(3) MDR as per Regulation (EU) 2023/607 are as follows:
- Class III and IIb implants: new transition period until 31 December 2027
- Class IIb and lower: new transition period until 31 December 2028
- Class III custom-made products: new transition period until 31 December 2026
- No ‘sell-off’ deadline
What does significant change mean?
A significant change in the design or intended purpose consists of two cumulative elements:
- There is a change in the design or intended purpose, and
- The change is significant
Therefore, changes that do not concern the design or intended purpose are out of scope of Article 120(3) of the MDR. Changes that concern the design or intended purpose only fall under Article 120(3) if they are considered ‘significant’.
Responsibilities of medical device manufacturers regarding ‘significance’ of changes
Medical device manufacturers have two primary responsibilities regarding changes:
- provide evidence and justification that a change does not affect the design or intended purpose, or
- in cases where the change affects the design or intended purpose of the device, that it is non-significant.
The outcome of this assessment should be documented and made available to a competent authority when requested.
If a change to a legacy device is not a significant change in design or intended purpose, it can be implemented using the above transitional arrangement. In these instances, manufacturers must adhere to the documentation criteria of AIMDD/MDD, meaning that the revised technical documentation must enable the evaluation of the product’s compliance with the relevant standards.
Determining a significant change to the design or intended purpose
The MDCG 2020-3 rev. 1 guidance document sets out whether a change in the design or intended purpose of a device is considered ‘significant’ within the meaning of EU MDR Article 120(3c), point (b).
Examples of changes in design and/or intended purpose that are ‘non-significant’:
- Changes related to corrective actions assessed and accepted by the competent authority
- Correction of spelling mistakes or merely editorial changes of the information to be supplied with the device (e.g. label or instructions for use)
- Clarifications of intended purpose, population, or clinical application in the information to be supplied with the device in line with the original certification
- Updates of the information to be supplied with the device (e.g. label, instructions for use or implant card) if they are required by EU legislation other than the MDR, are mere clarifications and do not adversely affect the devices’ safety and performance in relation to existing or new risks
The following diagram, taken from the MDCG 2020-3 rev. 1, helps understand when changes in design and/or intended purpose may be considered ‘significant’:
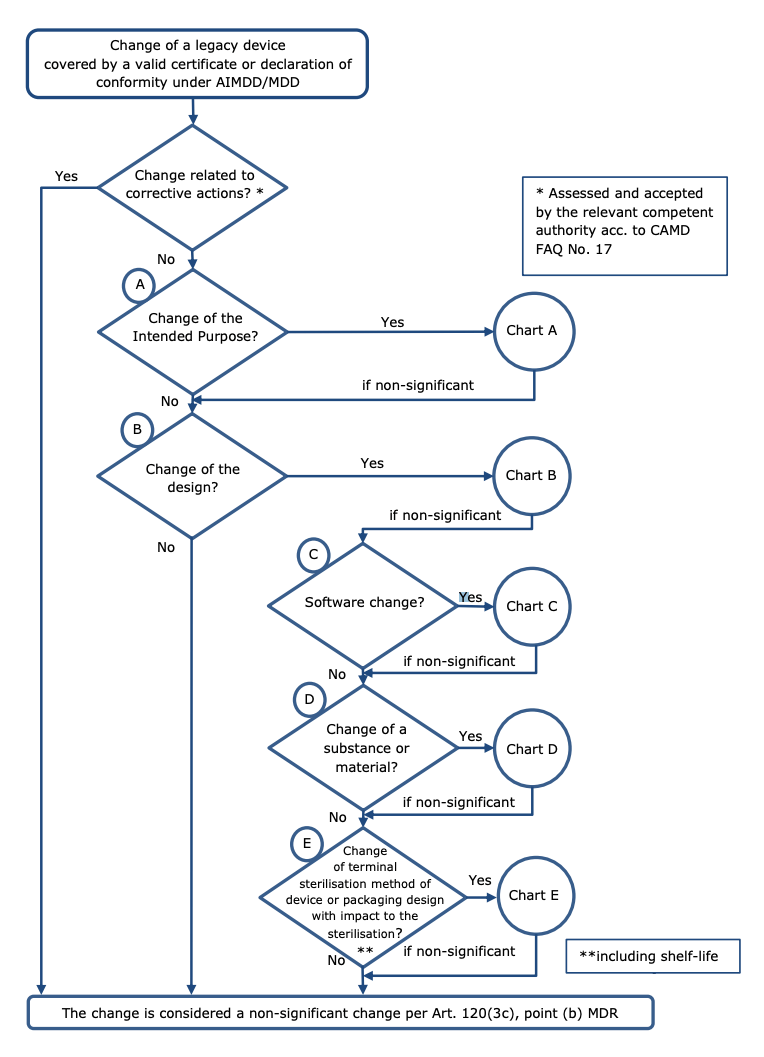
Charts A to E in the MDCG guideline contain specific areas where changes in the intended purpose, device design, device software, changes related to a substance or material or changes related to sterilisation are likely to be held to be significant. It’s vital to consult these charts when understanding whether a change will be significant and, by extension, whether your device can make use of the transitional arrangement.
In summary
The definition of a “significant change” involves two cumulative elements: a change in design or intended purpose that is deemed significant. Manufacturers bear the responsibility of determining and justifying whether a change in their medical device is significant or not, and therefore whether relevant technical documents require updating in line with the changes. The MDCG 2020-3 rev. 1 guidance document provides guidance on determining the significance of changes in design or intended purpose.
For further information regarding how we can meet your MDR requirements, including advice on whether changes are likely to be significant, please contact us for a free and confidential discussion.